PigVar
 Overview
Overview Pig SNPs
Pig SNPs SNP Table
SNP Table SNP Hit on Gene
SNP Hit on Gene SNP Coding
SNP Coding SNP lincRNA
SNP lincRNA Search SNPs by Region
Search SNPs by Region Browse on Chromosome
Browse on Chromosome SV
SV Positive Selection
Positive Selection Help
Help Manual
Manual Pipeline
Pipeline Data Statistics
Data Statistics Contact Us
Contact Us Download
Download
1. For the data from Ai et al. and Li et al., we used BWA mem (0.7.12-r1039) to align the Illumina short reads to the pig reference genome (Sus scrofa10.2). For the data generated by Wageningen University, the mapping files (bam files) were downloaded from European Nucleotide Archive (ENA) and realigned to the pig reference genome by BWA aln (0.7.5a-r405).
2. Use Picard (version: 1.119) to eliminate duplicated reads generated in library construction PCR.
3. Use tools in GATK (2.5-2-gf57256b) for each sample to realign reads around known indels, and recalibrate base quality score to get more accurate quality score for each base.
4. Use GATK UnifiedGenotyper to call a raw SNPs set from those refined data of all individuals, and then use the variant quality score recalibrate procedure in GATK to identify a high quality set of SNPs.
The directions (ancestral and derived) of the alleles for each SNP was deduced by comparison with outgroup species. Only SNPs agreed on the direction of the SNP in all the outgroup individuals were considered, with the criteria that the SNP needed to be sequenced in at least three individuals of the 7 outgroup individuals.
PigVar provides nucleotide diversity, Tajima’s D, Cockerham & Weir Fst, XP-EHH and XP-CLR scores for Chinese domestic pigs, Tibetan pigs and European domestic pigs. To facilitate the identification of true positive selection signals, users can use top 90% or 99% quantile in the calculated scores of selection distribution as cutoff.
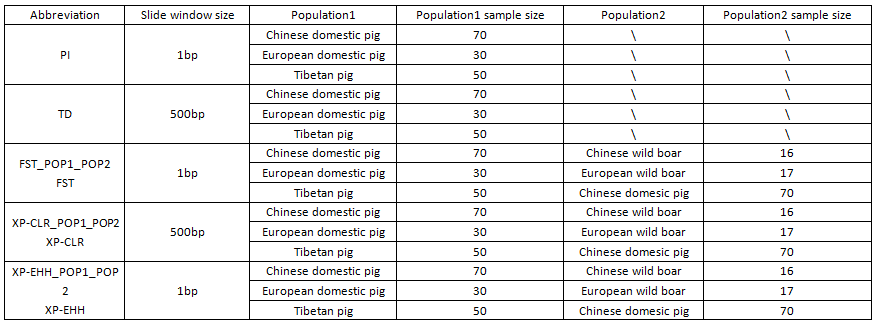
|
Kunming Institute of Zoology, CAS
and Xi'an University of Technology ©2014-2017 All Rights Reserved.
|


|